clover facilitates analysis and visualization of nanopore tRNA sequencing data, including differential expression, base-calling error analysis, and modification co-occurrence networks.
🚧🚧 clover is under active development. Caveat emptor. 🚧🚧
Installation
You can install the development version of clover from GitHub with:
# install.packages("pak")
pak::pak("rnabioco/clover")Example
clover reads output from the aa-tRNA-seq-pipeline and stores the results in a SummarizedExperiment.
library(clover)
library(SummarizedExperiment)
# Load pipeline results into a SummarizedExperiment
se <- create_clover(
config_path = clover_example("ecoli/config.yaml"),
sample_info = data.frame(
sample_id = c(
"wt-15-ctl-01",
"wt-15-ctl-02",
"wt-15-ctl-03",
"wt-15-inf-01",
"wt-15-inf-02",
"wt-15-inf-03"
),
condition = rep(c("control", "infected"), each = 3)
)
)
# Differential tRNA abundance with DESeq2
dds <- run_deseq(
assay(se, "counts"),
colData(se),
design = ~condition
)
res <- tidy_deseq_results(
dds,
contrast = c("condition", "infected", "control")
)
plot_volcano(res)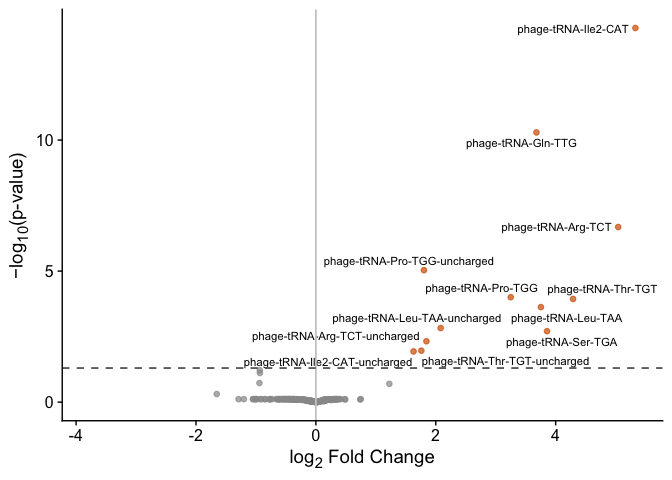
See vignette("clover") for a complete walkthrough.
Related work
- aa-tRNA-seq-pipeline is the Snakemake pipeline that generates the data clover analyzes.
- R2easyR visualizes structure probing signals on RNA secondary structure diagrams.
- nanoblot facilitates visualization of nanopore sequencing data, including a “virtual gel” plot.